We now have a new download site!
WGAViewer
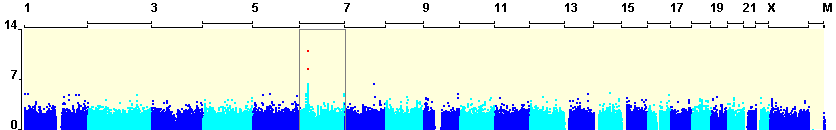
Dramatic advances in genotyping technologies have allowed the development of affordable products that simultaneously genotype a genome-wide set of polymorphisms that are known to represent most of the common genetic variants in specific human population groups. Because of these developments, the use of HapMap data (The International HapMap Consortium. 2007) and other genome resources has shifted from upstream SNP-selection tasks to the downstream tasks of interpreting the observed genotype-phenotype associations (Telenti and Goldstein 2006) . Ideally these resources should be used not only to help distinguish real associations from false positives ones, but should also help to generate hypotheses concerning the possible biological bases of observed associations.
These expectations create an immediate need to develop approaches that facilitate interpretation of a large set of P values in the context of known genomic features and also in the context of other studies of similar phenotypes. The ultimate goal is to allow investigators to consider the full set of P values resulting from an association study rather than simply looking at the few “top” polymorphisms with the lowest P values. This software package allows the researcher to visualize and consider other supporting evidence, such as the genomic context of the SNP, linkage disequilibrium (LD) with ungenotyped SNPs and the evidence from other Whole Genome Association projects alongside the P value of association when determining the potential importance of an individual SNP. Most importantly, it would highlight possible mechanisms, for example by directly or indirectly implicating a polymorphism with an apparent link to gene expression, splicing, non-coding RNAs or other possibilities that would suggest specific functional follow-up.
New! Extension to the WGAViewer program
Compared with previous discovery strategies, a whole-genome sequencing study is no longer constrained by differing patterns of linkage disequilibrium (Need and Goldstein, Trends in Genetics 2009;25(11):489-494), thus, in theory, is more possible to directly identify the gentic variants contributing to biological traits or medical outcomes.
The rapidly evolving high-throughput DNA sequencing technologies have now allowed the fast generation of large amount of sequence data for the purpose of performing such whole-genome sequencing studies, at a reasonable cost. SequenceVariantAnalyzer, or SVA, is a software tool that we have been developing to analyze the genetic variants identified from such studies.