1. Memory allocation
WGAViewer has some requirements on computer memory (RAM) to load the GWAS result sets and to perform the annotation tasks.
The default startup (which is also the minimum amount) will allocate up to 512MB of memory, which should be sufficient to load a genome-wide data file with up to 550K markers. A real time memory information bar monitors the memory allocation and usage (lower right corner, Figure 2.3-1). Green color indicates the memory allocation is adequate, while yellow or red indicates the program is running out of memory. In this case you may want to increase the default memory allocation by clicking on the memory monitoring bar (Figure 2.3-2). The program needs to be restarted to effect the change.
Usually the allocated memory should be at least greater than four-fold of the data file size, for example, if the data file is 30MB, the roughly estimated memory requirement should be at least 128M. This allocated number should not exceed the amount allowed by the system, that is, the “free” memory. Usually you may not allocate more than one half of your total memory. This means your computer should be equipped with at least 1-2GB RAM for a reasonable capacity and performance for a dataset including up to 550K markers.
In addition to this, Windows 32-bit version has certain rules of allocating memory to any user process, and these rules may affect the amount of memory you can allocate to the WGAViewer process (or any other user program). Our experience is that on a Windows 32-bit machine equipped with 3 GB of RAM, you may allocate up to 1.13 GB to the WGAViewer program. This amount of memory is sufficient to perform annotation for up to 600K markers.
For 'Windows Vista' users, there are some additional notes on 'virtual memory' specification.
We strongly recommend a 64-bit Windows or LINUX platform with at least 2-3 GB of free memory (that often means, 4-8 GB of RAM equipped) if you are going to work with datasets with 1 million or more markers, or to load multiple datasets simultaneously.
Lastly, for users who are working on a GWAS result set with a computer not sufficiently equipped, the following tip might be useful.
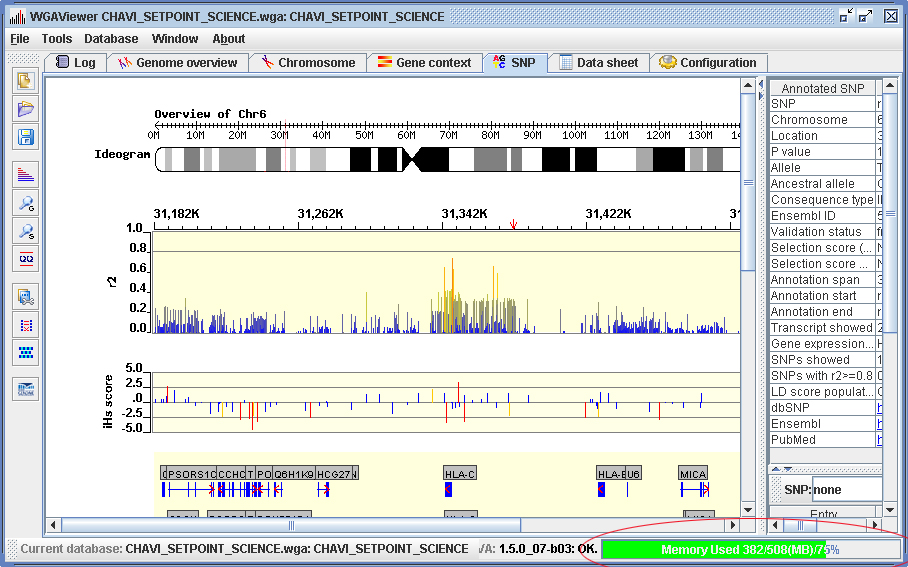
Figure 2.3-1 (Click to enlarge)
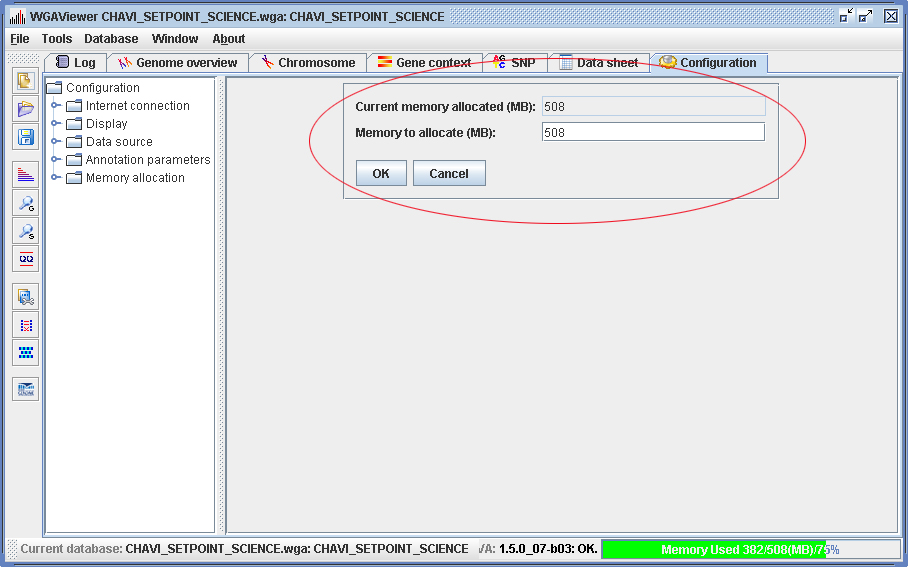
Figure 2.3-2 (Click to enlarge)
2. Tips to reduce memory requirement
You may create a subset of a WGAViewer flat-text data file (.wr) file, so that the requirement on memory can be significantly reduced. This subset is based on a P value criterion, for example, you may create a subset with all markers with P values less than 0.05.
To do this, click on menu "Tools -> Other tools -> Create a subset". Figure 2.3-3 shows an example. To load the generated subset, you only need around 50 MB of memory, instead of hundreds.
The main disadvantage of this trick is that you lose all the markers with P value greater than 0.05 (but of course the majority of which are not interesting anyway), and the marker density will be reduced significantly. In annotating such low-density datasets, especially in trying to locate genes, WGAViewer will automatically add markers, or 'anchors', at each side of the annotated region if needed, so that the mapping density can be maintained. The P values for these added 'anchors' will be set to -9 (missing).
Nevertheless, this trick might be useful for users who work with a computer not sufficiently equipped with memory.
Another trick you may want to play is that you may use some script to create chromosome-wise datasets and annotate them separately, especially after you already know where your top hits are located.
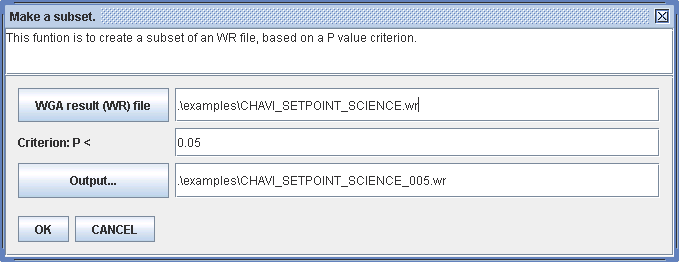
Figure 2.3-3 (Click to enlarge)