Top hits: Comprehensive annotation
In addition to these fast annotations, there is however one class of annotation that goes beyond convenient summaries of work that could be carried out manually and individually. The manual annotations using disparate databases are not feasible for any but the several top discoveries in any study. What happens with a polymorphism that only achieves a P value of 10 -5 , but which is itself in strong LD with an ungenotyped SNP that is annotated as definitely functional? This SNP would warrant special consideration, but would be missed in most manual settings. We have therefore established a slow annotation (typically 2-5 minutes per SNP) meant to pluck out such suggested associations and this is one of the few features of WGAViewer that can be viewed as moving toward real automated consideration of the full sets of results, as opposed to only expediting and summarizing analyses that would in any event have been completed. In addition to the gene context, LD extension, and expression annotation routines for each SNP, this process will also automatically check and filter the functions for both the original genotyped SNPs and their LD proxies. Once the annotation is done, the interactive filtering and other annotation features can be saved for convenient later use.
The questions that may be answered by this process include: what is the genomic context for each hit? What are those genes surrounding these hits? What are the SNPs and their P values around these hits? What is the LD context among these SNPs? How far does the LD extend for each hit? Do these hits represent proxies for functionally more relevant but ungenotyped variations? Do these hits or their proxies show any association with available functional data, for example, gene expression level? Etc.
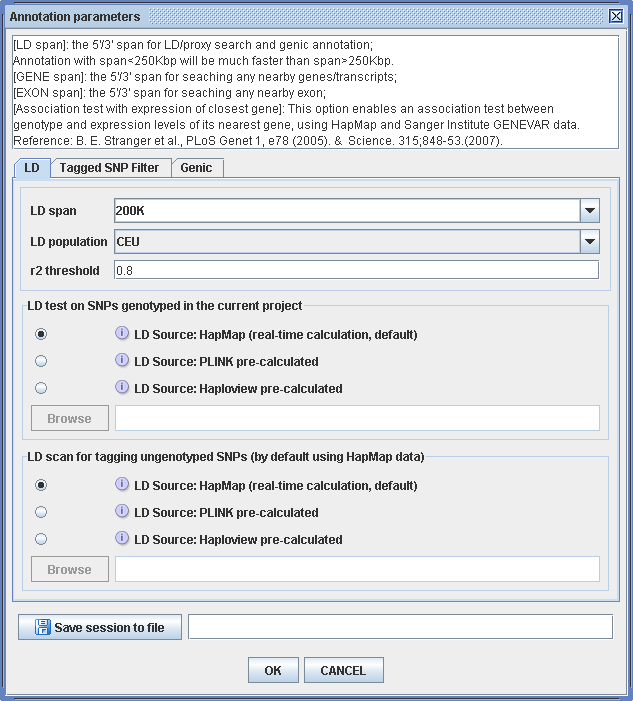
Figure 3.3.2-1. Parameters of comprehensive annotation for top hits. (Click to enlarge)
To perform this annotation, click on the blue hyperlink for each SNP rs# in Figure 3.3.1-2 individually, or click button “ Annotate all ” or “ Annotate remaining ” to launch a batch job. Either way will bring up a dialog for annotation parameters (Figure 3.3.2-1). The user then needs to specify the annotation parameters or leave the default values. “ LD span ” determines the main window size for the following annotation. “r2 threshold” will be used as the criterion to determine tagged SNPs for each hit. By default the LD test will be performed using HapMap data, but one has the options to use pre-calculated LD dataset as LD source , using either PLINK or Haploview. Gene span and exon span have the same definition as shown in figure 3.3.1-1, and will affect only those that have not been briefly annotated. The LD context is based on HapMap data. The specification of population and the cutoff for tagging can be setup in the “configuration” panel and will be discussed later in this documentation. If a batch annotation job is selected, an output file (.wga work session file) is mandatory. This is to avoid loss due to accidental interruption of the annotation procedure, for example, power surge, internet connection failure, etc. During this process WGAViewer will save the work session from time to time, so if the annotation is interrupted the saved work session can still be loaded and the annotation process can be restored by clicking on button “Annotate remaining” (Figure 3.3.1-2). Using the default parameters shown in Figure 3.3.2-1, around 2 minutes are required to annotate each SNP.
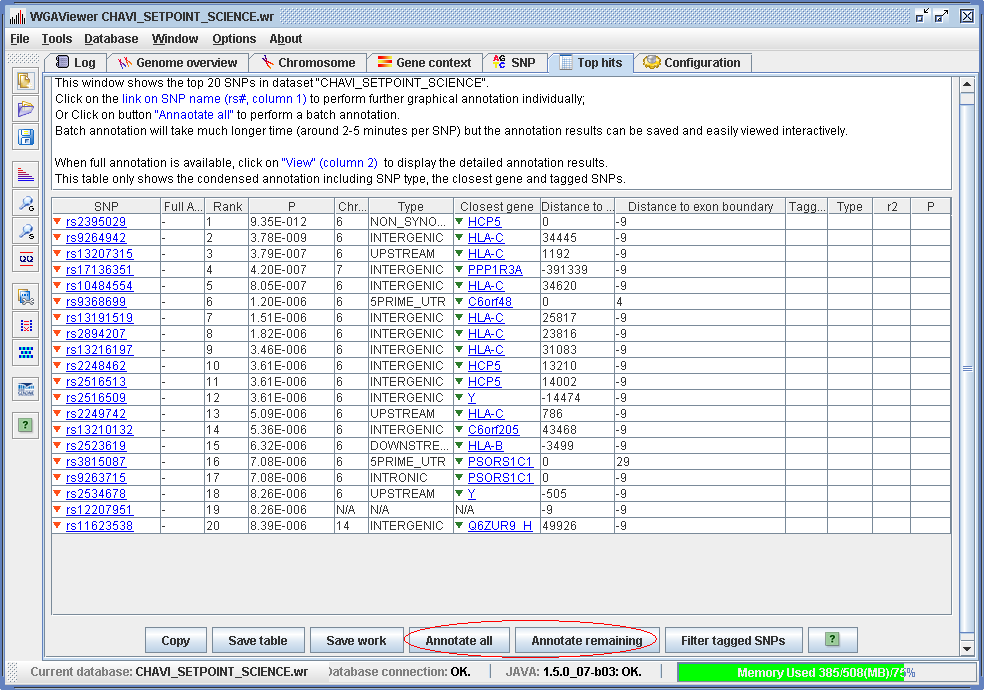
Figure 3.3.1-2. Results of first-round annotation for top hits. (Click to enlarge)
After the annotation is successfully performed (either individually or in a batch), a clickable “ View ” hyperlink will appear next to the column of rs# for each SNP with annotation results (Figure 3.3.2-2). Clicking on this “View” hyperlink will immediately bring up the saved annotation results with no further wait.
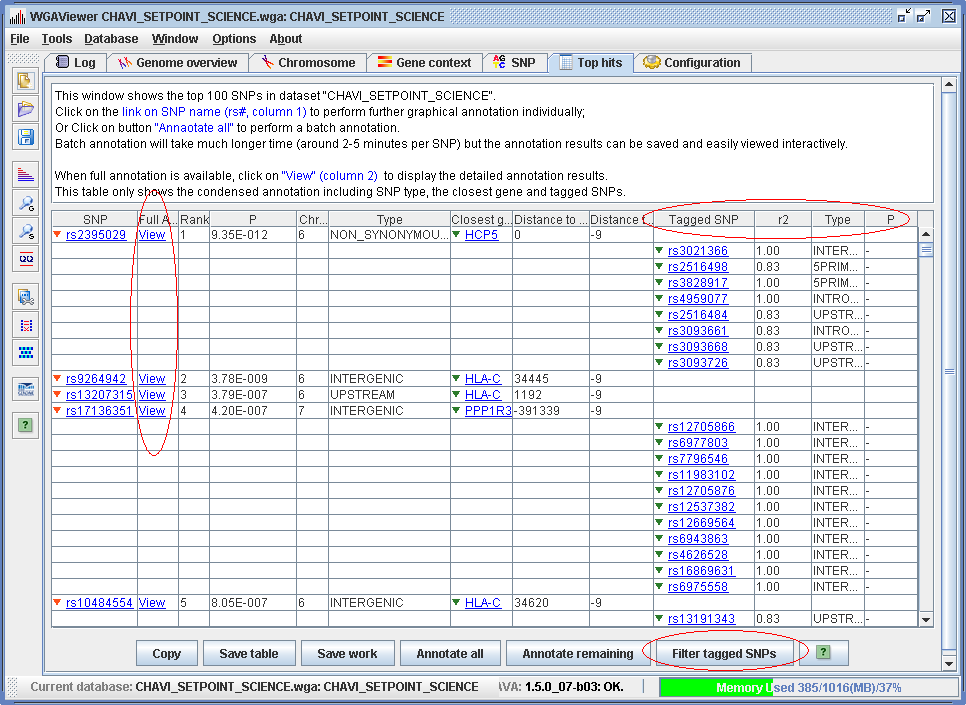
Figure 3.3.2-2. Results of comprehensive annotation for top hits. (Click to enlarge)
Like the resource menu for top hit and genes (Figure 3.3.1-3, 3.3.1-4), click on the tagged SNPs will bring up a resource menu which can direct to public databases (Figure 3.3.2-3).
{kind=link}
{kind=link}
Figure 3.3.2-3. Resource menu for tagged SNPs
The user also has the option to save the contents of this annotation summary table to a comma-separated text file (.CSV) by clicking on the “ Save table ” button, or to copy tab-separated data into the system clip board by clicking on the “ Copy ” button for pasting into any text editor or Microsoft EXCEL. To save the work session, click on button “ Save work ”. All the interactive features, annotation results can be reloaded from a saved work session (.wga file).
WGAViewer also offers an easy way to filter the SNPs tagged by the top hits. By clicking on button “ Filter tagged SNPs ” (Figure 3.3.2-2) the user can bring up a dialog for the filtering parameters (Figure 3.3.2-4). This dialog lists the 14 categories that are discussed in 3.3.1. This function provides a convenient data-mining method to screen the functionally relevant findings, for example, by excluding all the INTERGENIC HapMap SNPs that have been tagged by the user's WGA project.
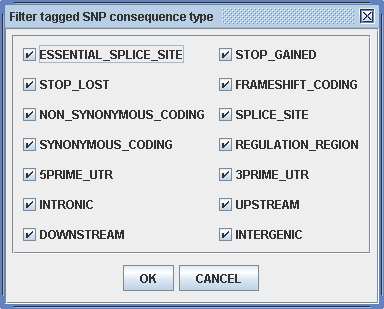
Figure 3.3.2-4. Filtering tagged SNPs.
As an illustration for this function we load the post-annotation example dataset by clicking on menu “File->Open an example dataset” and select “Post-annotation dataset” “CHAVI_SETPOINT_SCIENCE. wga”. Click on menu “Windows->Top hits viewer” to display the comprehensively annotated top 100 hits. Figure 3.3.2-2 shows the results. Different from the brief annotation results (Figure 3.3.1-2), the “Full annotation” column now has clickable hyperlinks “View”. And for some SNPs, a list of tagged SNPs and the type of the tagged SNPs are also shown in the table.
If one clicks on the hyperlink “View” (Figure 3.3.2-2), for example, on SNP rs9264942, the results of the comprehensive graphical annotation can then be shown sequentially. This involves the following three graphical panels.
Next: Top hits annotation: chromosome view; gene view; SNP view